Introduction
PrP is the only protein known to behave as a prion in animals, including humans. Present primarily in the nervous system[14], PrP is hypothesized to be the agent in a number of brain diseases infecting animals called transmissible spongiform encephalopathies (when examined microscopically, the brain appears to be ridden with tiny holes, hence appearing spongelike)[20]. Different misconfigurations of the PrP protein lead to different diseases, including scrapie in sheep and goats, bovine spongiform encephalopathy (commonly known as "Mad Cow Disease") in cattle, and Kuru and Creutzfeldt-Jakob disease in humans[20]. A number of prions also infect fungi.
Infection/Propogation
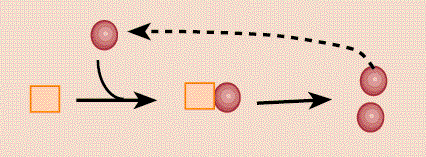
The simplest hypothesis for prion replication is that the misconfigured form simply binds to the normal form and converts it[15] (pictured). Prion diseases are peculiar since they can be acquired by infection, or inheritance, or develop spontaneously[13]. Hosts can easily go 10 years[4, p393] without developing symptoms, since presumably it would take a long time before one misfolded protein corrupts enough normal proteins to make the conversion process rapid.
References
[4]Campbell & Reece, Biology 8th Edition
[13]http://www.ibiology.org/ibioseminars/cell-biology/susan-lindquist-part-1.html
[14]http://en.wikipedia.org/wiki/PrP_structure
[15]http://www.nature.com/nature/journal/v389/n6653/fig_tab/389795a0_F2.html
[20]http://en.wikipedia.org/wiki/Transmissible_spongiform_encephalopathy
[13]http://www.ibiology.org/ibioseminars/cell-biology/susan-lindquist-part-1.html
[14]http://en.wikipedia.org/wiki/PrP_structure
[15]http://www.nature.com/nature/journal/v389/n6653/fig_tab/389795a0_F2.html
[20]http://en.wikipedia.org/wiki/Transmissible_spongiform_encephalopathy
